CASE REPORT
Hippokratia 2019, 23(3): 135-139
Toumba M1,2, Neocleous V2,3, Fanis P2,3, Tanteles GA3,4, Kyriakidou-Himonas M5, Picolos M6
1Pediatric Endocrine Clinic, IASIS Hospital, Paphos, 2Department of Molecular Genetics, Function & Therapy, The Cyprus Institute of Neurology & Genetics, Nicosia, 3Cyprus School of Molecular Medicine, Nicosia, 4Department of Clinical Genetics, The Cyprus Institute of Neurology & Genetics, Nicosia, 5Larnaca Endocrinology Center, Larnaca, 6Alithias Endocrinology Center, Nicosia, Cyprus
![]()
Abstract
Background: Resistance to thyroid hormone (RTH) is a rare dominantly inherited disorder mainly due to variants in the THRB gene leading to decreased end-organ responsiveness to thyroid hormones.
Case report: Clinical and molecular characteristics of four patients with RTH are described. Four patients with various phenotypes were studied; two prepubertal boys and two adults (one male and one female). Sequencing analysis of the THRB was performed. All individuals had persistently elevated free thyroxine and/or free triiodothyronine associated with non-suppressed thyroid-stimulating hormone (TSH), and all had non-autoimmune goiters of various sizes. In both adults, antithyroid drugs were previously administered without successful suppression of the thyroid hormones. The 27-year-old female had resting tachycardia as the only symptom. The 35-year-old male had a degree of cognitive impairment and was initially diagnosed with atrial fibrillation. The eight-year-old boy was diagnosed with attention deficit disorder and had resting tachycardia. The oldest boy (age nine years) underwent thyroid function tests as a part of the investigation for obesity and learning difficulties. Direct sequencing analysis of the THRB gene showed three previously reported variants: p.His435Leu (c.1304A>T) in the 35-year-old male, p.Pro453Thr (c.1357C>A) in the oldest boy, and p.Arg438Cys (c.1312C>T) variant in the other two patients.
Conclusions: Various phenotypes characterize common variants in the THRB gene, asymptomatic, thyroid hormone deprivation symptoms, or thyroid hormone excess symptoms. RTH should be suspected in both adults and children with elevated thyroid hormones and not suppressed TSH. A prompt molecular diagnosis and genetic counseling could prevent unnecessary tests and inappropriate treatments. HIPPOKRATIA 2019, 23(3): 135-139.
Keywords: Thyroid, resistance to thyroid hormone, THRB gene, free thyroxine, free triiodothyronine, FT4, FT3, thyroid stimulating hormone, TSH
Corresponding author: Meropi Toumba, MD, Pediatric Endocrine Clinic, IASIS Hospital, 8 Voriou Ipirou str., 8036, Paphos, Cyprus, tel: +35726848484, fax: +35726848300, e-mail: meropitoumba@gmail.com
Introduction
Resistance to thyroid hormone (RTH) syndrome is a dominantly inherited condition with an incidence of 1:40,0001. The condition results from pathogenic variants in genes that express the major thyroid hormone receptor isoforms, TRa, and TRb, located on the chromosomes 17 and 3, respectively. The synthesis of thyroid hormones is controlled by hypothalamic thyrotrophin-releasing hormone (TRH) and pituitary thyroid-stimulating hormone (TSH). Thyroid hormones T4 and T3 regulate TRH and TSH production by negative feedback. Mainly T3 acts by upregulating or downregulating the expression of thyroid receptor genes2,3.
Thyroid receptors have different tissue expression. TRa is most abundant in the central nervous system, myocardium, and skeletal muscle, while the TRb1 isoform is predominant in the liver and kidney, and the TRb2 isoform is most highly expressed in the pituitary and hypothalamus3. The phenotype varies depending on the degree of tissue responsiveness to elevated thyroid hormone levels3. A specific variant can result in various phenotypes in different patients, even within the same family4-8. The majority of the patients described in the existing literature are heterozygotes for variants in TRHB, whereas only a few patients with homozygous variants were described so far9. Patients with THRB variants present with RTH syndrome, which is typically characterized by elevated thyroid hormone levels, normal or mildly elevated TSH levels, goiter, and the absence of symptoms of thyrotoxicosis. Homozygote patients exhibit a more severe phenotype than heterozygote patients.
We report the first four cases with resistance to thyroid hormones syndrome in the small Mediterranean island of Cyprus. Although the identified mutations in our patients have already been described, it is important to report them considering the small population of the island, only 800,000 people.
Case report
The clinical and laboratory characteristics of the patients are summarized in Table 1.
Patient one
A 27-year-old female was self-referred for thyroid disease evaluation. She was under treatment for hyperthyroidism, diagnosed three years before. The patient had no significant past medical history; she was otherwise healthy with average body mass index (BMI) (23 kg/m2) and was complaining of occasional palpitations. Physical examination was unremarkable except for a mild goiter and slightly elevated resting heart rate of 100 pulse/min. While on propylthiouracil 50 mg twice daily, her thyroid function tests (TFTs) showed TSH: 3.46 (normal range: 0.27-4.2) mIU/ml, free thyroxine (FT4): 24.40 (12-22) pmol/l, and free triiodothyronine (FT3): 7.42 (3-6.5) pmol/l, whereas thyroid receptor antibodies were negative. Thyroid ultrasound was consistent with mildly enlarged thyroid without increased vascularity, and a very small right-sided nodule. The possibility of RTH was raised, and a molecular investigation was requested. The antithyroid drug was discontinued, and a repeated set of TFTs showed TSH: 0.85 mIU/ml, FT4: 25.04 pmol/l, FT3: 5.63 pmol/l. The patient’s father and two brothers also had elevated FT4 and non-suppressed TSH while her mother and sister had normal TFTs.
Initially, the proband was genotypically screened for the presence of variants at the THRB gene. Variants in this gene are known to be responsible for causing generalized thyroid hormone resistance characterized by goiter and high levels of circulating thyroid hormones (T3-T4), with normal or slightly elevated TSH. Sequencing analysis revealed that the proband was heterozygous for the previously identified p.Arg438Cys (c.1312C>T) THRB variant. Further DNA sequencing of the THRB gene revealed the variant p.Arg438Cys (c.1312C>T) in all the affected family members (Figure 1).
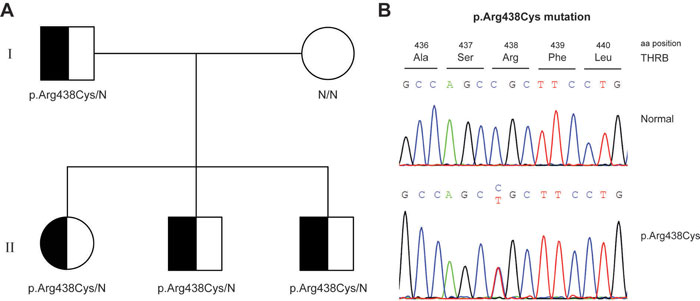
Figure 1: Genetic analysis of RTH syndrome: p.Arg438Cys variant in the family of patient one. A) Pedigree of the family of patient one. Squares and circles indicate males and females, respectively. Black shading indicates the presence of the p.Arg438Cys variant. B) Sequence electropherograms of the p.Arg438Cys variant.
Patient two
A 35-year-old male presented for evaluation of hyperthyroidism found as part of his work-up for atrial fibrillation. He had a history of palpitations. His atrial fibrillation was resolved and controlled with beta-blockers. On physical examination, he was clinically euthyroid with a small goiter and a mild degree of cognitive impairment. Otherwise, he was healthy but mildly overweight (BMI: 25.5 kg/m2). His initial TFTs were: TSH: 2.89 mIU/ml, FT4: 30.0 pmol/l, and FT3: 11.95 pmol/l for which he was already on treatment with carbimazole 5 mg twice a day. A new set of TFTs was requested while on the antithyroid drug, which showed TSH: 4.0 mIU/ml, FT4: 62.4 pmol/l, and FT3: 16.75 pmol/l. He also had negative thyroid-related autoantibodies. The thyroid ultrasound showed a homogeneous goiter with normal vascularity. Thyroid scintigraphy showed increased homogeneous isotope uptake. Magnetic resonance imaging (MRI) of the pituitary gland did not reveal any significant pathology. The serum alpha subunit was within normal limits. At this point, the suspicion of RTH was raised. Carbimazole treatment was discontinued, and THRB gene testing confirmed the presence of the already known p.His435Leu (c.1304A>T) variant (Figure 2). The patient’s thyroid function tests remained unchanged with elevated FT4 and FT3, and he is clinically stable and asymptomatic being only on low-dose of beta-blocker.

Figure 2: A) Sequence electropherograms of the identified p.His435Leu, and p.Pro453Thr variants in patients two and three, respectively. B) Schematic domain-structure diagram of THRB protein. The upper tier shows the exons corresponding to the THRB gene and the lower tier shows the structures of the THRB protein. The positions of the identified variants are indicated.
Patient three
A nine-year-old adopted boy was referred to the pediatric endocrinology clinic for obesity evaluation. On examination, his height was 144 cm and weight was 40kg both on the 90th centile and his BMI was 19 kg/m2 (75th centile). He had an enlarged goiter and tachycardia of 120 pulse/min. He was reported to have learning difficulties and attention deficit for which he was under the care of an educational psychologist. An IQ test was not available from his records, but he was managing well at school with average degrees. His thyroid hormones were significantly high (FT4: 54.90 pmol/l, FT3: 15.55 pmol/l) without having any symptoms of thyrotoxicosis. His TSH (2.56 mIU/ml) was not suppressed as expected. He did not take any medication. RTH syndrome was suspected, and molecular testing revealed p.Pro453Thr (c.1357C>A) variant on the THRB gene (Figure 2).
Patient four
An eight-year-old boy was referred to the clinical genetics department due to attention deficit and hyperactivity disorder and positive family history of RTH syndrome. His mother is the first cousin to patient one. At the age of seven years, he was diagnosed with attention deficit and hyperactivity disorder by a specialist pediatric psychiatrist, but he did not need any medication. Otherwise, he was developing well with unremarkable past history. His growth was normal at the 50th centile both for height (128 cm) and weight (26 kg) (BMI: 16 kg/m2, 50th centile). On clinical examination, he had resting tachycardia and a small goiter. His FT4 and FT3 were elevated (26.80 and 10.25 pmol/l, respectively), and TSH was in the normal range (1.89 mIU/ml). He did not take any treatment. DNA sequencing of the THRB gene revealed the same p.Arg438Cys (c.1312C>T) variant as on patient 1.
Direct sequencing of the THRB gene
Genomic DNA was isolated from peripheral blood leukocytes using a kit from QIAGEN (QIAGEN, GmbH D-40724, Hilden, Germany). The primers and conditions for polymerase chain reaction (PCR) amplification and direct sequencing of all 10 exons of the THRB (ENST00000396671.7) gene was performed using a set of primers as previously described10,11.
Discussion
In the present case study, we report three different known mutations on the THRB gene in four unrelated patients. Variants have been identified so far in over 300 families, most having single-nucleotide substitutions, resulting in one amino acid replacement and a few truncated molecules. Twenty other families have nucleotide deletions, insertions, or duplications, some producing frameshifts that create nonsense proteins. From the 171 different variants identified, the same variant may be carried by several different families. Diverse variants have resulted in different clinical manifestations6,11-13. The majority of the variants are of a single nucleotide substitution resulting in single amino acid replacement dominantly inherited12. All three variants described herein were previously reported13. Particularly p.Pro453Thr (c.1357C>A) was previously described in 26 unrelated families with similar phenotype13. Very rarely, other phenotypes may arise. Recently a patient with RTH due to p.Pro453Thr (c.1357C>A) variant was described to have also a thyrotropin secreting microadenoma14. It is not clear how variants to the THRB gene are related to thyrotropin secreting adenomas (TSHomas).
Patients with RTH present with variable phenotypes. Most of the individuals are completely asymptomatic, some manifest symptoms suggestive of thyroid hormone deprivation such as growth retardation, or/and impaired cognitive ability, while others show signs of thyroid hormone excess such as tachycardia, advanced bone age, or hyperactivity1-4. Not uncommonly, individuals have symptoms of both thyroid hormone deficiency and excess, such as the oldest boy in our study. As TRb2 isoform is most highly expressed in the pituitary and hypothalamus and TRa is most abundant in the central nervous system, myocardium and skeletal muscle an individual with THR syndrome may exhibit symptoms of hyperthyroidism in some tissues and hypothyroidism in other tissues depending on the degree of tissue responsiveness to elevated thyroid hormone levels3,4.
Interestingly, there is no specific correlation between phenotype and genotype. The same variant in the same family may be associated with thyrotoxic characteristics in some individuals or clinically euthyroidism in others with comparably elevated thyroid hormones. Other studies showed that the same variant had been associated with biochemical resistance in one patient or normal thyroid function tests in other members of the same family12-13. Similarly, in our report, the two related patients found with the same variant presented at a different age with different presenting symptoms. Goiter was a common symptom in the four patients; resting tachycardia characterized the three patients while one of the adults presented with atrial fibrillation. Notably, one of the boys was of normal growth and was diagnosed with attention deficit hyperactivity disorder and small goiter, while the other was obese with learning difficulties and enlarged thyroid. These different clinical findings suggest a marked phenotypic variability of this disorder, even in patients with the same variant.
There is no treatment available for RTH. The majority of individuals compensate by increasing the secretion of thyroid hormones. Patients with limited thyroidal reserve or simultaneous autoimmune thyroiditis may benefit from treatment with thyroxin. Additionally, patients with thyroid hormone excess symptoms may be helped by antianxiety drugs3,9.
Considering that the incidence of RTH due to variants in the THRB gene is very low6 and in the present study four index patients with three different variants were identified, it may be hypothesized that these variants are the result of ancestral variants that have spread over the Middle East area due to a possible founder effect. Similar founder effect phenomena have been anticipated for a number of other variants or diseases among the Middle East populations.
In conclusion, common variants in the THRB gene are characterized by various phenotypes and can be suspected in both adults and children with a different clinical and biochemical presentation. A prompt molecular diagnosis could prevent unnecessary tests and inappropriate treatments. More promising treatment options such as the development of thyroid hormone agonists and antagonists, allowing the blockage or the stimulation of TH on specific tissues are under investigation using animal models15.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Acknowledgments
1. The cases were reported on an e-poster presentation at the 57th ESPE Meeting – European Society for Paediatric Endocrinology, September 27-29, 2018, Athens, Greece; title: “Phenotype and genotype of four patients with thyroid hormone resistance syndrome due to mutations in the THRB gene” by Meropi Toumba, Vassos Neocleous, Pavlos Fanis, Nicos Skordis, Leonidas A Phylactou, George A Tanteles, Marinella Himonas, and Michalis Picolos. ePoster presentation.
2. We thank professor Nicos Skordis, MD, PHD (St George’s, University of London Medical School at the University of Nicosia, Cyprus and professor Leonidas A Phylactou, MD, PHD (Cyprus School of Molecular Medicine and The Cyprus Institute of Neurology and Genetics, Nicosia, Cyprus) for their comments and advice on the paper.
References
1. Lafranchi SH, Snyder DB, Sesser DE, Skeels MR, Singh N, Brent GA, et al. Follow-up of newborns with elevated screening T4 concentrations. J Pediatr. 2003; 143: 296-301.
2. Olateju TO, Vanderpump MP. Thyroid hormone resistance. Ann Clin Biochem. 2006; 43: 431-440.
3. Yen PM. Physiological and molecular basis of thyroid hormone action. Physiol Rev. 2001; 81: 1097-1142.
4. Paragliola RM, Lovicu RM, Locantore P, Senes P, Concolino P, Capoluongo E, et al. Differentiated thyroid cancer in two pa-tients with resistance to thyroid hormone. Thyroid. 2011; 21: 793-797.
5. Pappa T, Ferrara AM, Refetoff S. Inherited defects of thyroxine-binding proteins. Best Pract Res Clin Endocrinol Metabol. 2015; 29: 735-747.
6. Refetoff S, Dumitrescu AM. Syndromes of reduced sensitivity to thyroid hormone: genetic defects in hormone receptors, cell transporters and deiodination. Best Pract Res Clin Endocrinol Metabol. 2007; 21: 277-305.
7. Rivolta CM, Olcese MC, Belforte FS, Chiesa A, Gruñeiro-Papendieck L, Iorcansky S, et al. Genotyping of resistance to thyroid hormone in South American population. Identification of seven novel missense mutations in the human thyroid hormone re-ceptor beta gene. Mol Cell Probes. 2009; 23: 148-153.
8. Vinagre J, Borges F, Costa A, Alvelos MI, Mazeto G, Sobrinho- Simões M, et al. Differentiated thyroid cancer in patients with resistance to thyroid hormone syndrome. A novel case and a review of the literature. Front Mol Biosc. 2014; 1: 10.
9. Ono S, Schwartz ID, Mueller OT, Root AW, Usala SJ, Bercu BB. Homozygosity for a dominant negative thyroid hormone receptor gene responsible for generalized resistance to thyroid hormone. J Clin Endocrinol Metabol. 1991; 73: 990-994.
10. Sørensen HG, van der Deure WM, Hansen PS, Peeters RP, Breteler MM, Kyvik KO, et al. Identification and conse-quences of polymorphisms in the thyroid hormone receptor alpha and beta genes. Thyroid. 2008; 18: 1087-1094.
11. Lado-Abeal J, Dumitrescu AM, Liao XH, Cohen RN, Pohlenz J, Weiss RE, et al. A de novo mutation in an already mutant nucleotide of the thyroid hormone receptor beta gene perpetuates resistance to thyroid hormone. J Clin Endocrinol Metab. 2005; 90: 1760-1767.
12. Hamon B, Hamon P, Bovier-Lapierre M, Pugeat M, Savagner F, Rodien P, et al. A child with resistance to thyroid hor-mone without thyroid hormone receptor gene mutation: a 20-year follow-up. Thyroid. 2008; 18: 35-44.
13. Guo QH, Wang BA, Wang CZ, Wang M, Lu JM, Lv ZH, et al. Thyroid hormone resistance syndrome caused by heter-ozygous A317T mutation in thyroid hormone receptor β gene: Report of one Chinese pedigree and review of the literature. Medicine (Baltimore). 2016; 95: e4415.
14. Teng X, Jin T, Brent GA, Wu A, Teng W, Shan Z. A Patient With a Thyrotropin-Secreting Microadenoma and Re-sistance to Thyroid Hormone (P453T). J Clin Endocrinol Metab. 2015; 100: 2511-2514.
15. Flamant F, Samarut J. Thyroid hormone receptors: lessons from knockout and knock-in mutant mice. Trends Endo-crinol Metab. 2003; 14: 85-90.